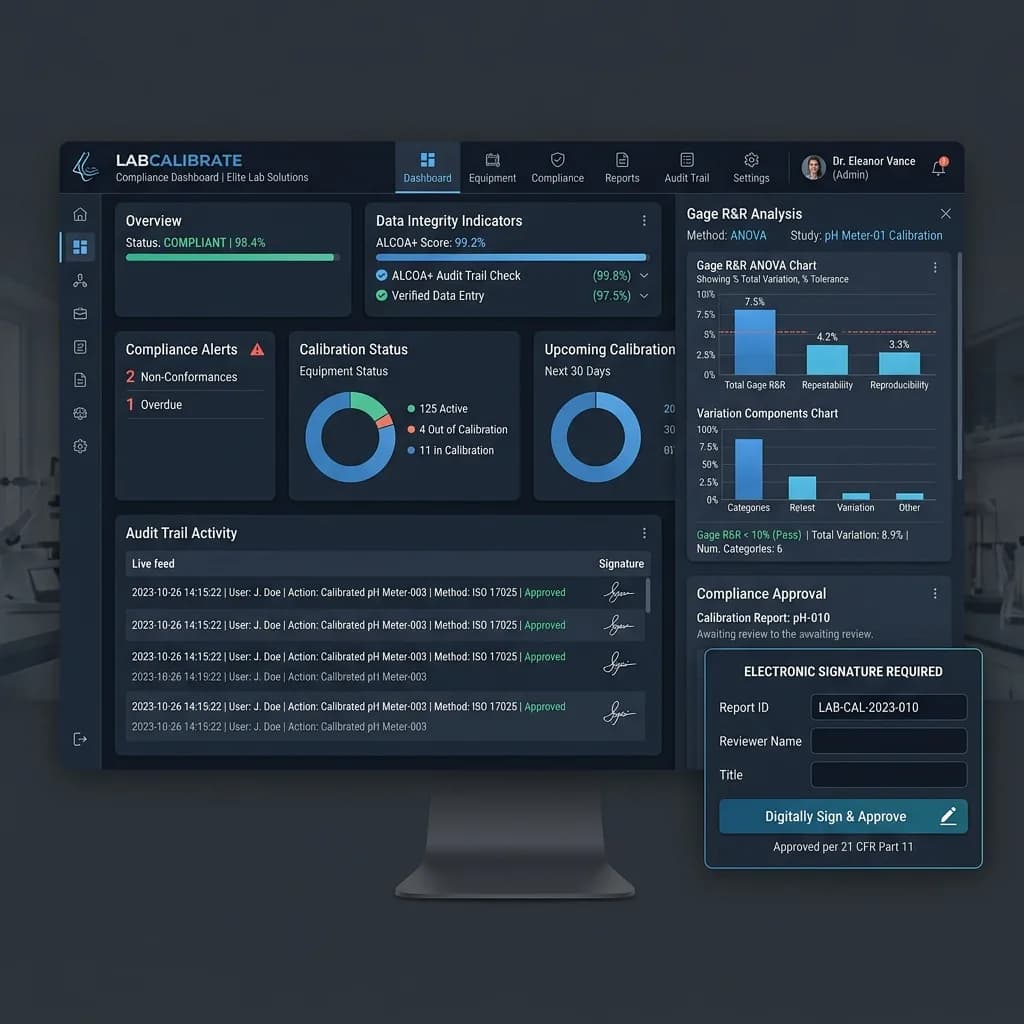
Executive Summary
Why Spreadsheet-Based Calibration Creates Systematic Compliance Risk Spreadsheet-based calibration management consistently fails three critical requirements of 21 CFR Part 11, ISO 17025, and IATF 16949: it does not produce a tamper-evident audit trail; it does not support compliant electronic signatures; and it is structurally vulnerable to data integrity issues that regulatory authorities have elevated as their primary inspection focus.
What a Compliant Calibration Programme Actually Requires Requirements differ by framework — pharmaceutical manufacturers face different obligations than automotive Tier 1 suppliers. ISO 9001, IATF 16949, ISO 17025, GMP, and FDA 21 CFR Part 11 each impose specific calibration management requirements mapped against functional system requirements in this paper.
How to Transition Without Disrupting Operations The fear of disruption is the most common reason calibration improvements are deferred. A structured transition approach using data migration, parallel running, and phased cutover achieves full transition in 6 to 8 weeks without disrupting ongoing calibration activities.
1. Calibration Management — A Compliance Obligation
1.1 The Measurement Dependency of Quality Systems Every quality system in manufacturing depends, ultimately, on measurement. Products are accepted or rejected based on measurements. Process parameters are controlled based on measurements. Release decisions are made based on measurements. The compliance evidence that regulatory authorities review is built from measurements. And the reliability of every measurement depends on the calibration status of the instrument that took it.
An analytical balance used to weigh an API dose that is 2% out of calibration does not produce weights that are 2% wrong — it produces weights that are systematically biased in a direction and by an amount that the analyst does not know is wrong. Every dosing decision made with that balance is compromised by an unknown error.
Calibration management is a compliance obligation because the entire quality system depends on measurement reliability — and measurement reliability depends on calibration.
2. The Global Data Integrity Enforcement Context
Data integrity has emerged as the dominant regulatory enforcement theme in pharmaceutical manufacturing globally over the past decade. FDA warning letters citing data integrity failures have increased substantially. MHRA, WHO, and other regulatory authorities have published specific data integrity guidance documents and made data integrity assessment a standard part of GMP inspections.
Indian pharmaceutical manufacturers have been disproportionately represented in FDA data integrity enforcement actions. Several high-profile warning letters issued against Indian facilities specifically cite calibration management failures — expired calibrations used for measurements, records backdated or modified, and systems that did not produce reliable, tamper-evident records.
2.1 The ALCOA+ Data Integrity Framework The ALCOA+ principles provide the foundation for data integrity assessment: • Attributable: Every calibration entry linked to an identified technician • Legible: Digital records eliminate handwriting legibility issues • Contemporaneous: Data entered at calibration bench, not transcribed later • Original: Electronic records must be originals, not paper copies • Accurate: System controls must prevent modification after completion • + Complete: Mandatory fields enforced by system; no missing data • + Enduring: Systematic retention with defined retention periods • + Available: Instant retrieval — especially during regulatory inspections
3. Calibration Requirements by Regulatory Framework
ISO 9001:2015 — Clause 7.1.5 Instruments must be calibrated at specified intervals against traceable standards. Every instrument must be identified to enable calibration status determination. Records retained as documented information. Out-of-tolerance instruments must have actions recorded.
IATF 16949:2016 — Clause 7.1.5.1 and 7.1.5.2 Adds Measurement System Analysis (MSA) requirements. Gage R&R studies required for measurement systems affecting product quality. Customer notification required when suspect product may have been shipped due to an out-of-calibration instrument. MSA results retained as documented information per AIAG MSA-4.
ISO 17025:2017 — Laboratory Accreditation Measurement results must be traceable through an unbroken calibration chain with defined measurement uncertainty. Calibration certificates must include uncertainty statements. Calibration personnel must have documented competency assessments.
GMP / cGMP — Pharmaceutical Manufacturing Analytical instruments must be qualified (IQ/OQ/PQ). Calibration at defined intervals with documented justification. Out-of-calibration impact assessment required — scope of affected products must be documented. Records retained for shelf life plus one year or regulatory mandate, whichever is longer.
FDA 21 CFR Part 11 — Electronic Records and Signatures System-generated, tamper-evident audit trails. Electronic signatures with printed name, date/time, and meaning. System access controls. Operational system checks enforcing permitted step sequences. Applies when electronic records replace paper in FDA-regulated pharmaceutical and medical device manufacturing.
4. The Spreadsheet Problem — Specific Compliance Failures
Spreadsheet-based calibration management systems present critical vulnerabilities when evaluated against regulatory standards:
• System-generated audit trail (21 CFR Part 11 / GMP): FAILS — Excel has no tamper-evident audit trail. This is an automatic regulatory finding in 21 CFR Part 11 inspections. • Compliant e-signatures (21 CFR Part 11): FAILS — typed names are not compliant signatures. Records are not equivalent to controlled approvals. • Contemporaneous entry (ALCOA+): AT RISK — transcription from paper is not contemporaneous, causing data integrity vulnerability where the original record is unclear. • Access controls (21 CFR Part 11): WEAK — shared file access bypasses password protection, leading to unauthorised access and modification risk. • Deletion traceability (All frameworks): FAILS — records can be permanently deleted without trace. It is impossible to demonstrate that records were not removed. • MSA integration (IATF 16949): NOT AVAILABLE — requires separate disconnected tools. MSA evidence is disconnected from the calibration record history.
5. What a Compliant Calibration Management System Must Provide
Instrument Registry with Complete Attribute Capture Every instrument registered with identifying information, measurement category, location, calibration frequency, and current status. Status — Calibrated, Due, Overdue — maintained automatically from scheduled dates and calibration events.
Automated Scheduling and Pre-Due Alerts Calibration intervals defined per instrument. Due dates calculated automatically. Alerts distributed to technicians and quality managers at configurable lead times. The system functions without manual intervention regardless of personnel changes.
Calibration Records with ALCOA+ Properties Records created at the time of the activity, by the identified technician, with server-generated timestamps, protected from modification after closure. As-found and as-left values, reference standard used, and result recorded per instrument.
Tamper-Evident System-Generated Audit Trail Every action on every record logged with user identity, timestamp, and before/after content of any change. Read-only to all users. System-generated, not user-maintained. Available as a report for regulatory review.
Compliant Electronic Signatures Identity-authenticated, non-transferable signatures with required fields — printed name, date/time, meaning of signature — meeting 21 CFR Part 11 technical requirements. GAMP 5 validation documentation available.
Out-of-Tolerance Workflow Automatic detection. Instrument status changed to restricted. Non-conformance workflow initiated. Impact assessment documentation supported — scope of affected measurements since last valid calibration documented and actionable.
6. Measurement System Analysis — The IATF 16949 Dimension
Calibration and MSA address different but related aspects of measurement system quality. Calibration addresses systematic error — bias. MSA addresses random error — variation across repeated measurements, operators, and conditions. IATF 16949 requires both. The AIAG MSA-4 reference manual defines acceptance: %R&R less than 10% is generally acceptable; 10 to 30% may be acceptable depending on application; above 30% requires investigation and improvement.
PrecisionCAL includes MSA and Gage R&R study functionality as a built-in capability — not a separate tool or spreadsheet. Results stored against the instrument record, available for IATF documentation alongside calibration history.
7. Multi-Site Calibration Management — The Enterprise Challenge
Managing calibration across multiple sites manually or with siloed systems creates significant visibility and consistency issues:
• Overdue instrument visibility: In a siloed system, each site checks its own records with no consolidated view. An enterprise platform provides a live dashboard showing all statuses in real time across all sites. • Cross-site benchmarking: Manual compilation leads to inconsistent formats. An enterprise system automates this, presenting the same metrics across all sites consistently. • Regulatory inspection prep: Manual compilation takes 3–5 days per inspection. An enterprise system allows all records to be generated via report in 2–4 hours. • Personnel change resilience: In manual systems, knowledge and access are lost when individuals leave. An enterprise platform ensures scheduling and alerts continue regardless of personnel changes.
8. Transition From Manual Systems — A Structured Approach
Phase 1 — Data Migration (Weeks 1–3) Instrument registry and historical calibration records migrated from existing spreadsheet system. PrecisionCAL's migration concierge handles this import — no manual re-entry, historical record maintained in the new platform.
Phase 2 — Schedule Configuration (Week 3–4) Calibration intervals, alert recipients, and approval workflows configured to match existing programme. Configuration reflects current requirements rather than designing a new programme from scratch.
Phase 3 — Parallel Running (Weeks 4–7) Both systems running simultaneously. Validates that the new system captures the same information as the existing one. Builds team confidence before full cutover. No disruption to ongoing calibration activities or production.
Phase 4 — Cutover and Decommission (Week 7–8) Spreadsheet decommissioned. New platform is the system of record. Historical records in the system from migration. Typical timeline: 6 to 8 weeks from initiation to full operation.
9. The Business Case for Calibration Management Investment
The business case for a compliant calibration management system is typically driven by risk avoidance and operational efficiency:
• FDA 483 observation (calibration finding): Can lead to ₹50 lakhs to ₹5 crores+ in costs due to consulting, CAPA, reinspection, and potential import alert impacts. • IATF 16949 calibration non-conformance: Results in ₹5–50 lakhs in follow-up audit costs, certification risk, and customer notification requirements. • Product recall linked to out-of-cal instrument: Can cost potentially crores. The recall scope is determined by traceability quality; poor records equal a wider recall scope. • Audit prep (current vs system-based): Takes 60–70 hours manually versus 4–8 hours with a system. This represents significant time recovered at quality personnel rates across every audit.
The cost of an enterprise licence is typically a fraction of the first avoided compliance event.
10. Conclusion
Calibration management is the foundation of measurement reliability — and measurement reliability is the foundation of every quality system in regulated manufacturing. The current state of calibration management in Indian pharmaceutical and automotive manufacturing — predominantly manual, spreadsheet-based, and structurally non-compliant with applicable regulatory requirements — represents a systematic compliance risk that is avoidable.
The path to a compliant, sustainable calibration programme is not technically complex. It requires moving from a system that tracks calibration to a system that manages it — with automated scheduling, at-source digital record capture, tamper-evident audit trails, and compliant electronic signatures. The investment is modest relative to the cost of the compliance failures it prevents. For manufacturers in regulated markets, it is not an optional enhancement. It is a baseline requirement.

